Phonon Structure, Infra-Red and Raman Spectra of Li$_2$MnO$_3$ by First-Principles Calculations
Abstract
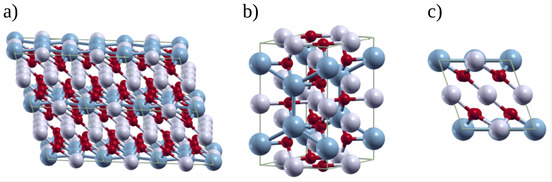
The layer-structured monoclinic Li$_2$MnO$_3$ is a key material, mainly due to its role in Li-ion batteries and as a precursor for adsorbent used in lithium recovery from aqueous solutions. In the present work, we used first-principles calculations based on density functional theory (DFT) to study the crystal structure, optical phonon frequencies, infra-red (IR), and Raman active modes and compared the results with experimental data. First, Li$_2$MnO$_3$ powder was synthesized by the hydrothermal method and successively characterized by XRD, TEM, FTIR, and Raman spectroscopy. Secondly, by using Local Density Approximation (LDA), we carried out a DFT study of the crystal structure and electronic properties of Li$_2$MnO$_3$. Finally, we calculated the vibrational properties using Density Functional Perturbation Theory (DFPT). Our results show that simulated IR and Raman spectra agree well with the observed phonon structure. Additionally, the IR and Raman theoretical spectra show similar features compared to the experimental ones. This research is useful in investigations involving the physicochemical characterization of Li$_2$MnO$_3$ material.
Full citation
R. Pulido, N. Naveas, R. J. Martin-Palma, F. Agulló-Rueda, V. R. Ferró, J. Hernández-Montelongo, G. Recio-Sánchez, I. Brito, and M. Manso-Silván,
“Phonon Structure, Infra-Red and Raman Spectra of Li$_2$MnO$_3$ by First-Principles Calculations,”
Materials. 15, 6237 (2022).
DOI: 10.3390/ma15186237